
Sudden Cardiac Death In Young Athletes

Abstract
Sudden cardiac death (SCD) in an athlete is a rare yet highly visible tragedy that generates significant media attention and discussion among medical personnel, sports communities, and laypersons alike. The incidence of SCD is greater in athletes compared with their nonathletic counterparts due to the increased risk associated with strenuous exercise in the context of a quiescent cardiac abnormality. Numerous structural, electrical, and acquired cardiovascular abnormalities are capable of causing SCD, many of which can be identified during life and managed by lifestyle modifications, pharmacotherapy, and device therapy. Strategies for the prevention of SCD, including pre-participation cardiovascular screening, are endorsed by sports governing bodies, but mandatory pre-participation cardiovascular screening remains rare. Evaluation of athletes poses diagnostic difficulties, particularly differentiating between physiological adaptation to exercise, known as athlete's heart, and cardiomyopathic processes capable of causing SCD. This paper provides a detailed review regarding the etiology of SCD in young athletes and provides insight into the challenges and dilemmas faced when evaluating athletes for underlying pathological conditions.
The beneficial effects of regular exercise for primary and secondary prevention of cardiovascular disease are well documented (1). Paradoxically, athletes harboring quiescent cardiovascular abnormalities are at greater risk of exercise-related sudden cardiac death (SCD). Data from Italy have shown a 2.8-fold greater risk of SCD among competitive athletes compared with their nonathletic counterparts. SCD results from intense physical exercise in the context of an underlying cardiovascular abnormality (2). The mechanism is typically ventricular arrhythmia, probably due to exercise-induced catecholamine surges acting on an arrhythmogenic substrate. Postulated contributory mechanisms also include dehydration, hyperpyrexia, electrolyte imbalances, and increased platelet aggregation associated with exercise (3).
The precise definition of an athlete is complex; however, for the purposes of this review, we define an athlete as an individual engaged in regular physical training and participating in official sports competition with an emphasis on excellence and achievement (4). SCD in a young athlete is often difficult for the public to comprehend because athletes are perceived as the healthiest segment of society. Most nontraumatic deaths are attributed to cardiovascular abnormalities that can be identified during life and managed with lifestyle modifications including abstinence from exercise of high or moderate intensity, pharmacotherapy, and implantable cardioverter-defibrillators (ICDs) (5). Prevention of such catastrophes by pre-participation cardiovascular screening (PPS) is recommended by learned organizations and sports governing bodies including the European Society of Cardiology (ESC), American Heart Association, and the International Olympic Committee (6–8).
This paper provides a detailed review regarding the etiology of SCD in young athletes and provides insight into the challenges and dilemmas faced when evaluating athletes for underlying pathological conditions.
Epidemiology of SCD in Athletes
The incidence of SCD among young athletes is a source of debate, particularly as studies differ in their methodology. Data compiled retrospectively from media reports, insurance claims, and electronic databases are likely to represent a significant underestimate. The most robust data are derived from prospective, observational studies in Italy using regional registry data with mandatory reporting systems, which report incidence rates of 3.6/100,000 per year in the pre-screening era. More recent cross-sectional studies from the United States have demonstrated relatively similar figures with incidence rates ranging from 2.3 to 4.4/100,000 per year (9–11).
There is a significant male predominance of SCD among athletes. Data from the National Center for Catastrophic Sports Injury Research on high school and college athletes reported a 5-fold higher incidence of SCD in male compared with female athletes (12). In the Veneto region of northern Italy, where >110,000 athletes were evaluated over a 21-year follow-up period, the incidence rates of SCD were 2.6/100,000 person-years in male athletes and 1.1/100,000 person-years in their female counterparts (2). Several factors are implicated in this sex difference including lower participation rates among female athletes at the elite level, although this trend is changing, and lower prevalence of cardiac abnormalities capable of causing SCD in females (13).
Over the past 3 decades, there has been an explosion in the number of African/Afro-Caribbean (black) athletes competing at the elite level (14). SCD appears to be more common in this ethnic group, with a reported incidence rate of 5.6/100,000 per year in the United States (11). Cardiomyopathy has been consistently demonstrated as the most common cause of exercise-related SCD in young athletes. Data from U.S. autopsy series have reported higher death rates from hypertrophic cardiomyopathy (HCM) in black compared with white athletes (20% vs. 10%, respectively), raising concern that this condition may exhibit a more malignant phenotype in black individuals (15).
Sudden death occurs more frequently in certain sports. In the United States, basketball and football have the greatest incidence, whereas in Europe, soccer predominates (16). Extrapolation of this observation suggests that individuals participating in sports of high dynamic and low isometric intensity are at higher risk of death. However, there is the potential for data bias due to higher participation rates in these sports.
Etiology of SCD in Athletes
In athletes older than 35 years of age, 80% of SCD is frequently due to atherosclerotic coronary artery disease, but in younger athletes, inherited and other acquired cardiovascular abnormalities are usually responsible (Fig. 1). Cardiomyopathy, including HCM and arrhythmogenic right ventricular cardiomyopathy (ARVC), is the most common cause of exercise-related SCD (5).
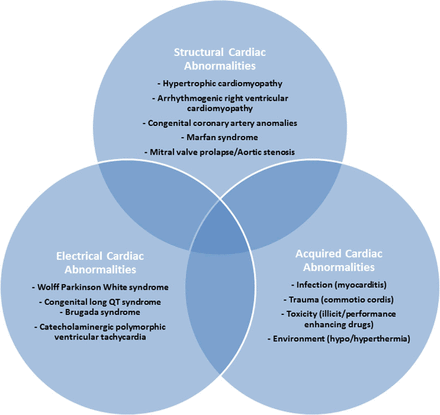
Figure 1: Common Causes of Sudden Cardiac Death in Young Athletes - The common causes of SCD in young athletes <35 years old can be divided into structural, electrical, and acquired cardiac abnormalities (22).
Structural cardiac abnormalities
Hypertrophic Cardiomyopathy
The reported prevalence of HCM is 0.2% in the general population and 0.07% to 0.08% in athletes (17). HCM is a primary myocardial disorder with an autosomal dominant pattern of inheritance, characterized by left ventricular hypertrophy (LVH) in the absence of abnormal loading conditions and myocardial disarray on histology. Sudden death due to ventricular tachycardia (VT)/ventricular fibrillation (VF) is often the first clinical manifestation (18). Deaths caused by HCM are common in start-stop sports, for example, football and basketball, but rare in endurance events such as rowing, long-distance cycling, and running. It is hypothesized that the combination of myocardial hypertrophy, impaired myocardial relaxation, myocardial ischemia, and dynamic left ventricular (LV) outflow obstruction impede augmentation of stroke volume for prolonged periods, and individuals with HCM are therefore usually selected out of endurance sports.
The diagnosis is made using electrocardiography (ECG) and echocardiography. More than 90% of affected individuals have an abnormal resting electrocardiogram (Figs. 2A and 2C) (19). All individuals with previous cardiac arrest or sustained VT are at high risk of SCD and require treatment with an ICD. Other recognized risk factors for SCD include: 1) unheralded syncope; 2) family history of SCD; 3) severe LVH (>30 mm); 4) sustained or nonsustained VT; and 5) attenuated blood pressure response to exercise. These 5 risk factors have low positive predictive value but high negative predictive value. Subjects exhibiting ≥1 of the 5 risk markers should be considered for prophylactic insertion of an ICD (20).
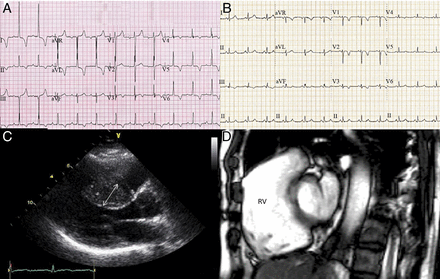
Figure 2: ECG and Imaging Abnormalities Observed in HCM and ARVC - (A) Electrocardiogram (ECG) in hypertrophic cardiomyopathy (HCM) demonstrating left axis deviation, voltage criteria for left atrial enlargement, and left ventricular hypertrophy. There is ST-segment depression with deep T-wave inversion in leads I, aVL, and V5 and V6, and T-wave inversion in leads II and V3 and V4. (B) ECG in arrhythmogenic right ventricular cardiomyopathy (ARVC) demonstrating T-wave inversion in right precordial leads. (C) Transthoracic echocardiogram demonstrating HCM with significant asymmetrical septal hypertrophy (arrow) measuring 22 mm. (D) Cardiac magnetic resonance scan showing ARVC with right ventricular (RV) dilation and wall fibrosis.
Arrhythmogenic Right Ventricular Cardiomyopathy
ARVC has a reported prevalence of 1/1,000 in the general population. It is an inherited myocardial disease caused by mutations in genes encoding cardiac desmosomal proteins (21). The mechanism of SCD is complex and myocardial stretch and myocyte detachment during exercise are thought to result in ventricular arrhythmia and SCD. In survivors, focal myocarditis with subsequent healing leads to progressive fibrofatty replacement of the myocardium and a propensity to VT/VF. Macroscopic appearances include right ventricular (RV) dilation, dysfunction, and aneurysm formation (22). Exercise exacerbates these pathophysiological changes, and a 5-fold higher risk of SCD in ARVC has been demonstrated during competitive sports compared with sedentary activity (2). Diagnosis relies on meeting the 2010 ARVC Task Force criteria, which include symptoms, family history, resting/ambulatory electrocardiographic changes, echocardiographic and cardiac magnetic resonance imaging and myocardial tissue characterization (Figs. 2B and 2D) (23). Previous cardiac arrest, unexplained syncope, VT with hemodynamic compromise and extensive structural disease including LV involvement are risk factors for SCD and should prompt consideration of prophylactic ICD implantation (20,24).
Congenital Coronary Artery Anomalies
Congenital coronary artery anomalies (CCAAs) reportedly cause SCD in 12% to 33% of athletes. The most common anomalies implicated are left coronary artery origins in the right sinus of Valsalva and right coronary artery origins in the left sinus of Valsalva (25). SCD results from ventricular arrhythmia triggered by myocardial ischemia during exercise. Coronary blood flow is impaired by the abnormal ostium of the anomalous vessel, compression of the anomalous artery as it courses between the pulmonary artery and ascending aorta, and/or coronary spasm triggered by endothelial dysfunction (26). Victims of SCD due to CCAA are often asymptomatic before presentation, although chest pain associated with syncope should raise suspicion of the disorder.
Diagnosis using ECG, echocardiography, and exercise stress testing is notoriously difficult because affected individuals rarely reveal features of inducible ischemia during exercise stress testing or pharmacological functional tests. Cardiac magnetic resonance angiography and computed tomography coronary angiography are the gold standard imaging modalities (27). The usual recommended therapy for CCAA is surgical correction; however, there is controversy regarding which specific types of CCAA require surgical correction in an asymptomatic athlete. Although surgery is almost universally recommended for left-sided CCAA, clinical management of a right-sided CCAA is more uncertain. It has been suggested that an intramural course, acute-angled take off, stenosis, or slitlike opening carry higher risk (25).
Other Structural Cardiac Abnormalities
Other structural cardiac abnormalities associated with SCD include aortic dissection/rupture typically in the context of Marfan syndrome, mitral valve prolapse (MVP), and aortic stenosis. Marfan syndrome is a collagen disorder caused by mutations in the gene encoding fibrillin, inherited as an autosomal dominant trait with a prevalence of 1 in 5,000. It accounts for approximately 3% of exercise-related SCD in young athletes and is characterized by skeletal, cardiac, and ocular abnormalities. Cystic medial necrosis in the tunica media of the aorta results in aortic dilation, dissection, or rupture, which may be expedited by the increases in aortic pressure associated with exercise (28,29). Marfan patients should be prohibited from isometric or isotonic exercise of moderate to high intensity. Individuals with an enlarged aortic root (>40 mm) should receive a beta-blocker to help retard aortic dilation (30).
MVP affects 3% to 5% of the general population; however, <100 cases of SCD have been reported in which MVP was the only abnormality identified, and only 3 occurred during physical exertion. Most individuals are asymptomatic. In rare instances, the condition is associated with VT, although the exact mechanism is unknown. The relatively high-frequency of MVP in the general population raises the question of whether identification of MVP in a victim of SCD is causal or coincidental (31,32). In general, athletes with MVP are allowed to continue to compete; however, competitive sport is precluded when MVP is associated with moderate to severe mitral regurgitation, severe chest pain, exertional syncope, documentation of VT, a long QT interval, or Marfan syndrome (30).
Aortic stenosis due to a congenital bicuspid aortic valve is a rare but recognized cause of SCD in young athletes that can be identified through basic screening efforts involving cardiovascular physical examination. Athletes with mild aortic stenosis may compete in low- to moderate-intensity dynamic or static sports provided that they are asymptomatic and free of documented arrhythmia, with normal LV function both at rest and during exercise echocardiography (30).
Electrical cardiac abnormalities
Wolff-Parkinson-White Syndrome
Wolff-Parkinson-White syndrome describes ventricular pre-excitation due to anterograde conduction via an accessory atrioventricular pathway with paroxysmal arrhythmias that usually result from atrioventricular re-entrant tachycardia (33). The prevalence of pre-excitation in athletes is similar to that in the general population (0.1% to 0.3%) and may be revealed by a delta wave, short PR interval, and prolonged QRS duration on the electrocardiogram (Fig. 3A). Most affected athletes are asymptomatic, but some report symptoms of palpitations. If atrial fibrillation develops in individuals with Wolff-Parkinson-White syndrome, there is a risk of SCD from VF secondary to rapid anterograde conduction via the accessory pathway (34). Determining the electrical properties of the accessory pathway is therefore crucial for establishing the risk of SCD. Curative catheter ablation of the accessory pathway in adults permits return to competitive sport after 3 months (33). Recent guidance suggests that in adolescents, only high-risk pathways require catheter ablation; however, referral to an electrophysiologist is recommended (35).
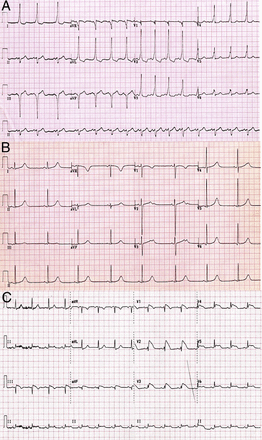
Figure 3: Electrocardiographic Patterns Observed in Electrical Cardiac Abnormalities Associated With Sudden Cardiac Death - (A) Wolff-Parkinson-White syndrome with characteristic short PR interval, delta wave, prolonged QRS interval, and ST-segment depression in leads I, aVL, and V2 through V4. (B) Congenital long QT syndrome with QTc interval of >460 ms. (C) Brugada syndrome with characteristic coved-type and down-sloping ST-segment elevation in leads V1 through V4 (type 1 electrocardiographic pattern).
Congenital Long QT Syndromes
Long QT syndrome (LQTS) comprises a group of hereditary ion channelopathies with a prevalence of 1 in 2,000 to 5,000 in the general population. The incidence of SCD in athletes due to LQTS was 2% in the U.S. registry data (15). Although 12 different culpable genes have been identified, >70% of cases are due to loss-of-function mutations in KCNQ1 (encoding potassium channel IKs; LQT1) and KCNH2 (encoding potassium channel IKr; LQT2) and gain-of-function mutations in SCN5A (encoding sodium channel INa; LQT3). Abnormal cardiac repolarization predisposes to polymorphic VT/VF. Individuals present with pre-syncope, syncope, palpitations, or SCD. LQTS may be misdiagnosed as epilepsy due to myoclonic movements occurring during syncope. LQT1 is most commonly associated with SCD during exercise, in particular, swimming and diving, likely due to adrenergic surges that occur with sudden immersion in cold water (36).
LQTS should be considered when the QTc interval exceeds 440 ms in males or 460 ms in females, in the absence of medications capable of causing acquired QT interval prolongation (37). Athletes may exhibit slightly longer QT intervals compared with the general population and 0.4% to 0.7% of highly trained athletes may exhibit values >440 to 460 ms (see the following text). The 36th Bethesda Conference guidelines therefore recommend restriction of athletic individuals with a QTc interval exceeding 470 ms in males and 480 ms in females to improve the positive predictive value (29). The diagnosis of LQTS should be made using probability scoring systems that include symptoms, family history, electrocardiographic changes (Fig. 3B) and evidence of torsade de pointes, as some affected individuals have a QTc interval within the normal range (38). High-risk features include a QTc interval >500 ms and adolescents or females with the LQT2 genotype. Individuals with a history of aborted SCD, recurrent syncope, and polymorphic VT despite medical therapy with beta-blockers should be considered for prophylactic implantation of an ICD (39).
Brugada Syndrome
Brugada syndrome (BrS) is an autosomal dominant sodium channelopathy with an incidence of 1 in 2,000 to 5,000 (40). The condition is characterized by a partial right bundle branch block pattern on the electrocardiogram with associated coved ST-segment elevation (BrS type 1 electrocardiographic pattern) (Fig. 3C). Individuals present with syncope or SCD due to polymorphic VT/VF. Mixed phenotypic expressions of the disease, ranging from distinct repolarization abnormalities to subclinical cardiac conduction defects, also occur. BrS is not typically associated with exercise-related SCD; however, increased vagal tone induced by chronic athletic conditioning at rest and exercise-induced hyperthermia may enhance the propensity to SCD and may trigger ventricular arrhythmias (41). The only established treatment is ICD insertion. Although deaths from BrS characteristically occur at rest, intensive exercise is generally not advised because it may be associated with profound bradycardia and core temperatures exceeding 40°C, both of which may precipitate fatal arrhythmias in affected individuals.
Catecholaminergic Polymorphic VT
Catecholaminergic polymorphic VT is associated with mutations in the ryanodine receptor, calsequestrin, and ankyrin-B proteins, which predispose to adrenergically mediated polymorphic VT and recurrent syncope provoked by physical exercise or emotional stress (42). Typically, the baseline electrocardiogram is unremarkable, but exercise stress testing may provoke multifocal ventricular premature beats or VT with beat to beat 180° alternating QRS axis (bidirectional VT). The disorder commonly presents in adolescence, and affected individuals may have a family history of juvenile sudden death or stress-induced syncope. Prevention of SCD includes medical therapy with beta-blockers and ICD insertion in those who continue to experience symptoms despite medical therapy.
Acquired cardiac abnormalities
Commotio Cordis
Blunt trauma to the chest can trigger VF and SCD without causing direct injury to the thoracic cage or heart (43). Commotio cordis typically occurs in sports with projectile objects such as ice hockey, lacrosse, and baseball. High-risk contact sports such as martial arts and player collisions in team sports such as soccer have also been implicated. Commotio cordis is more common in children and adolescents due to a thin and compliant thoracic cage that allows greater transmission of energy to the heart. In animal models, a single blow directly over the precordium 10 to 30 ms before the T-wave peak on the electrocardiogram has been demonstrated to induce VF. A rapid increase in LV pressure follows, which appears to activate ion channels via mechanoelectric coupling, resulting in the generation of an inward current, augmentation of repolarization, and nonuniform myocardial activation. Subsequent premature ventricular depolarizations trigger VF and SCD (44).
Myocarditis
Myocarditis, typically caused by viral infections (e.g., Coxsackie B) accounts for up to 7% of SCD in athletes (45). The diagnosis of myocarditis should be considered in any healthy young individual with recent viral illness, new exercise intolerance, clinical signs of cardiac failure, electrocardiographic repolarization abnormalities, and/or echocardiographic regional wall motion abnormalities. Active myocarditis can be identified on cardiac magnetic resonance imaging. Acute illness is associated with ventricular arrhythmia. In some cases, myocarditis can lead to a dilated cardiomyopathy with ongoing symptoms of cardiac dysfunction and increased risk of SCD (46). Athletes diagnosed with myocarditis should refrain from sports activity for a 6-month convalescent period to reduce the risk of SCD.
Performance-Enhancing Drugs
The use of performance-enhancing drugs occurs in athletes, although the true incidence is unknown. Anabolic-androgenic steroids, stimulants such as ephedrine, and nonsteroidal agents such as recombinant human erythropoietin have been associated with SCD, but a causal relationship is difficult to ascertain given that their use is forbidden. Anabolic androgenic steroids have been shown to change lipoprotein metabolism leading to premature atherosclerosis and myocardial infarction. These agents also induce hypertension and both anabolic-androgenic steroid and ephedrine use may result in cardiomyopathy and ventricular arrhythmias. Toxicological investigation is therefore recommended after a SCD event in an athlete (47).
Premature Coronary Artery Disease
In large series from Italy and the United States, premature atherosclerotic coronary artery disease accounted for 2% to 3% of SCD in young athletes (9,15). It is most commonly a manifestation of familial hypercholesterolemia. Symptoms are rare, and SCD is often the first presentation; however, peripheral stigmata including xanthelesma, corneal arcus, and xanthomata are more common and should raise suspicion.
Evaluation of Athletes and Diagnostic Dilemmas
PPS is supported by sports organizations at the international and national levels (6). However, a potential limitation is the need for highly trained medical personnel with experience evaluating athletes, interpreting their clinical data, and organizing relevant further investigations when required without affecting training regimens and sporting events. An algorithm for the assessment of athletes for conditions capable of causing SCD is presented in Figure 4.
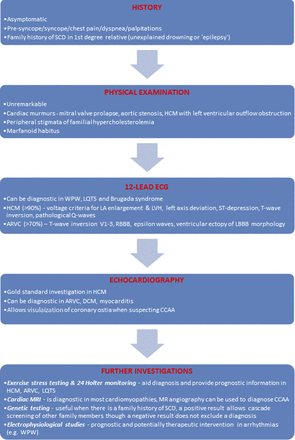
Figure 4: Algorithm for Evaluating Athletes for Conditions Capable of Causing SCD - The evaluation of athletes for conditions predisposing to SCD must incorporate clinical history, physical examination, 12-lead ECG and trans-thoracic echocardiography with further investigations as required. CCAA = congenital coronary artery anomaly; DCM = dilated cardiomyopathy; LA = left atrium; LBBB = left bundle branch block; LQTS = long QT syndrome; LVH = left ventricular hypertrophy; MRI = magnetic resonance imaging; RBBB = right bundle branch block; SCD = sudden cardiac death; WPW = Wolff-Parkinson-White syndrome.
Diagnostic Dilemmas
Athlete's heart
Regular physical exercise can lead to physiological adaptation in cardiac dimensions, including increased LV wall thickness (LVWT) and LV cavity size, which may be reflected on ECG and echocardiography (48). Such remodeling, termed athlete's heart, permits enhanced filling of the left ventricle in diastole and augmentation of stroke volume allowing generation of a large and sustained cardiac output even at rapid heart rates. The magnitude to which this adaptation occurs is influenced by several demographic factors including age, sex, body surface area, sport undertaken, and ethnicity (3). Consequently, a significant diagnostic dilemma can arise when attempting to differentiate physiological adaptation, with associated electrocardiographic and echocardiographic changes, from cardiac pathology. This is particularly important because false-positive diagnoses may lead to erroneous disqualification from a sport with loss of earnings and significant psychological distress to the athlete, whereas false-negative evaluations may result in devastating SCD.
The ESC has devised recommendations for electrocardiographic interpretation in athletes, which have reduced false-positive rates, in Caucasian athletes at least. Group 1 changes result from physiological adaptation of the cardiac autonomic nervous system and occur in as many as 80% of athletes, but Group 2 changes occur in <5% and are suggestive of underlying cardiovascular disorders, most notably cardiomyopathy and ion channelopathy.
Athlete's heart versus HCM
A proportion of male athletes, predominantly those involved in endurance sports, demonstrate extreme physiological adaptation with LVWT measurements of 13 to 15 mm (49). Although the majority of individuals with HCM have a mean LVWT of 18 to 20 mm, ∼8% have morphologically mild hypertrophy. Therefore, a male athlete with an LVWT of 13 to 15 mm falls into a ‘grey zone’, where differentiation between physiological LVH and HCM is crucial. This diagnostic dilemma occurs in 2% to 4% of white male athletes and 12% to 18% black male athletes (Fig. 5) (50,51).
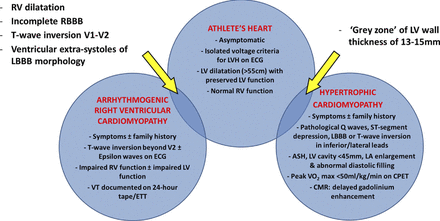
Figure 5: Differentiating Between Physiology and Pathology: ‘Athlete's Heart’ Versus HCM and ARVC - Regular exercise can lead to physiological adaptation of cardiac structure and function (athlete's heart) which can be identified by changes on ECG and echocardiography. There is some overlap with HCM and ARVC (yellow arrows). Key features can be used to differentiate between physiology and pathology. ASH = asymmetrical septal hypertrophy; CMR = cardiac magnetic resonance imaging; CPET = cardiopulmonary exercise test; ETT = exercise tolerance test; LV = left ventricular; RV = right ventricular; VT = ventricular tachycardia; other abbreviations as in Figures 2 and 4.
In the majority of athletes, differentiating physiology from pathology is possible using ECG and echocardiography. Isolated QRS voltage criteria for LVH are commonly observed in athletes but occur in only 2% of individuals with HCM. Electrocardiographic changes suggestive of HCM include T-wave inversion, pathological Q waves, ST-segment depression in >2 contiguous leads, and left bundle branch block. Inverted T waves >1 mm in depth in ≥2 contiguous leads except V1 and V2 in white athletes and V1 through V4 in black athletes should raise suspicion for HCM (52). Physiological LVH is homogeneous and associated with chamber enlargement and normal indexes of diastolic function. In contrast, individuals with HCM often show asymmetrical patterns of LVH, small chamber size, and impaired diastolic function. End-diastolic LV dimensions >55 mm are common in trained athletes but are rare in HCM in which the LV cavity size is usually <45 mm. In addition, HCM is associated with abnormal pulsed and tissue Doppler indexes of LV diastolic filling and impaired relaxation. In selected athletes, cardiac magnetic resonance imaging may offer incremental diagnostic value for detection of segmental LVH in the anterolateral free wall, posterior ventricular septum or apex, and demonstration of delayed gadolinium enhancement indicative of myocardial fibrosis (53). In equivocal cases, the presence of a family history of HCM or SCD and low peak oxygen consumption (peak Vo2max <50 ml/kg/min) on cardiopulmonary exercise testing favor a diagnosis of HCM. Genetic analysis has a high positive predictive value but a low negative predictive value and remains costly and time-consuming (54). In rare cases, re-evaluation with ECG and echocardiography after a period (8 to 12 weeks) of detraining may be the only practical method of differentiating between the 2 entities.
Athlete's heart versus ARVC
The diagnosis of ARVC in athletes is particularly challenging. Early in the disease process, the “concealed phase,” the heart may appear morphologically normal (Fig. 5). Minor electrocardiographic abnormalities in RV leads, infrequent ventricular extra systoles of RV origin, and subtle changes in the right ventricle may be the only objective manifestations of the disorder, and these may overlap with physiological adaptation of the right ventricle to regular exercise (22). The presence of epsilon waves, abnormal late potentials on signal averaged ECG, nonsustained VT of left bundle branch block morphology, and RV regional wall motion abnormalities favor the diagnosis of ARVC (55).
The majority of current data regarding RV size and function are determined on the basis of dimensions derived from small cohorts of normal individuals. Previous imaging studies have demonstrated that athlete's heart represents balanced cardiac enlargement with proportional increases in LV and RV mass, LV and RV end-diastolic volumes, and LV and RV stroke volumes compared with sedentary controls (56). The impact of exercise on the right ventricle has not been studied as comprehensively as the left ventricle, probably due to difficulties arising from its complex shape and trabeculated structure. However, studies of endurance athletes have shown that cardiac remodeling is not limited to the left heart. Data from 102 subjects demonstrated chronic RV cavity enlargement with RV outflow tract values greater than the proposed major criteria for ARVC in 28% of endurance athletes (23,57). As-yet unpublished data from our group have demonstrated significantly increased echocardiographic RV dimensions in professional soccer players, greater than the upper reference limits defined by the ESC and American Society of Echocardiography (58). This increases the difficulty in differentiating between physiological adaptation and ARVC, and larger studies are necessary to determine physiological upper limits of RV dimensions in athletes.
QTc interval in athletes
The diagnosis of congenital LQTS is determined on the basis of a triad of a prolonged QTc interval, unheralded syncope or documented polymorphic VT, and family history of SCD or LQTS. Although the prevalence of LQTS is 1 in 2,500 to 10,000 in the general population, in athletes, QTc interval prolongation is reportedly as high as 0.4% to 0.7% (equivalent to 1 in 150 to 250) and therefore higher than the prevalence of cardiomyopathy (59). Current data on SCD in young athletes indicate that death due to ion channel disease occurs in 2% to 4% of cases (15,16). The relatively high prevalence of QTc interval prolongation in athletes and the low death rate of LQTS suggest that the majority of causal mutations may be relatively benign and/or that most athletes with an isolated long QTc interval do not actually have LQTS. Isolated QTc interval prolongation in an athlete may represent delayed repolarization as a result of increased LV mass, autonomic adaptation, or the fact that Bazett's formula undercorrects the QTc interval at low heart rates (59). However, as many as 34% of SCDs in athletes from the U.S. registry data remain unexplained and undiagnosed ion channel disease may therefore also account for a proportion of this group (15). Our own data, which was determined on the basis of 118 consecutive SCD referred to a tertiary cardiac pathology center, failed to identify an abnormality in 23% of cases. However, it could be argued that the high incidence of normal hearts may represent a referral bias because deaths associated with structurally normal hearts were more likely to be referred to a tertiary cardiac pathology center for more detailed assessment (60).
Black athletes
Studies have demonstrated that black individuals exhibit more marked electrocardiographic repolarization changes and a greater magnitude of LVH on echocardiography compared with white individuals (14). It can be extrapolated that exercise-associated increases in preload and afterload may result in greater physiological cardiac adaptation in black compared with white athletes, leading to greater overlap with disease phenotypes, in particular HCM.
Data from the United Kingdom have demonstrated significantly greater LVH by voltage criteria (68% vs. 40%, respectively; p < 0.001), repolarization abnormalities including ST-segment elevation (85% vs. 62%, respectively; p < 0.001), and deep T-wave inversion (12% vs. 0%, respectively; p < 0.001) in black athletes compared with their white counterparts (51). T-wave inversion occurs predominantly in electrocardiographic leads V1 through V4. Further comprehensive clinical evaluation and follow-up data has shown no evidence of underlying cardiomyopathy in black athletes with anterior T-wave inversion in leads V1 through V4, suggesting that this is likely to represent a benign finding in this ethnic group (Figs. 6A and 6B).
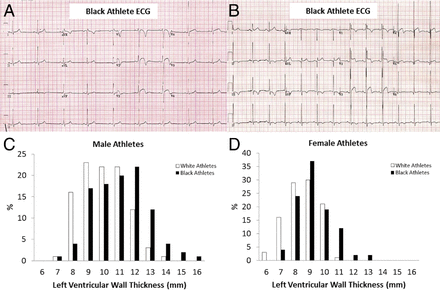
Figure 6: Patterns of Repolarization Changes and Distribution of LVWT in Black Male and Female Athletes and Their White Counterparts - (A) Electrocardiogram (ECG) of a black male marathon runner demonstrating ST-segment elevation in leads V1 through V3 with deep T-wave inversion consistent with physiological adaptation to exercise in this ethnic group. (B) ECG of a black male boxer demonstrating ST-segment elevation in leads V1 through V4 but associated left ventricular hypertrophy and deep T-wave inversion in leads II, III, and aVF, suggestive of cardiac pathology. (C) Distribution of left ventricular wall thickness (LVWT) (in millimeters) in black male and white male athletes demonstrating a significant proportion of black male athletes with a maximal LVWT between 13 and 16 mm, compatible with morphologically mild hypertrophic cardiomyopathy (HCM). (D) Distribution of LVWT in black female and white female athletes also demonstrating a significant proportion of black female athletes with a maximal LVWT ≥12 mm, compatible with morphologically mild HCM.
Echocardiographic data from athletes matched for age, body surface area, and sport showed significantly greater LVWT in black than white athletes (11.3 mm vs. 10.0 mm, respectively; p < 0.001) (Fig. 6C) (51). Among black athletes, 18% exhibited an LVWT ≥13 mm, and 3% had an LVWT ≥15 mm, in the range consistent with morphologically mild HCM. In contrast, only 4% of white athletes had an LVWT ≥13 mm, and none had an LVWT ≥15 mm. None of the black athletes with LVH exhibited other phenotypic features of HCM on echocardiography, cardiac magnetic resonance imaging, exercise stress testing, or 24-h electrocardiographic monitoring. In black male athletes, an LVWT ≥13 mm warrants further investigation but may represent physiological adaptation, whereas an LVWT ≥16 mm is suggestive of underlying pathology. The greater prevalence of repolarization changes on ECG and increased LVWT on echocardiography have also been demonstrated to a lesser extent in female (Fig. 6D) and adolescent black athletes (61,62).
Early repolarization in athletes
Early repolarization (ER) was historically considered a benign electrocardiographic variant but has emerged as a risk marker for SCD. Inferior/inferolateral ER with a horizontal/descending ST-segment is associated with idiopathic VF in the general population (63). The prevalence of ER is higher in athletes, ranging from 22% to 43% and is more common in male black athletes with lower heart rates and greater training duration (64). It is typically observed in the lateral electrocardiographic territory with rapidly up-sloping ST-segment morphology. The majority of evidence suggests that ER in athletes is benign, although 1 study has shown an increased prevalence of inferior/inferolateral ER with a horizontal/descending ST-segment in athletic survivors of cardiac arrest (65). At present, there is insufficient evidence to make recommendations for competitive sports participation.
Primary and Secondary Prevention of SCD in Athletes
Pre-participation cardiovascular screening
PPS of athletes is recommended by both the American Heart Association and ESC. In the United States, a 12-point screening protocol encompassing symptoms, family history, and physical examination is recommended, whereas in Italy, a mandatory state-sponsored screening program incorporating 12-lead ECG, symptoms, family history, and physical examination exists for all competitive athletes (5). The Italian PPS protocol has been in place for >25 years, and on the basis of the data generated from this experience, the ESC recommends PPS including ECG in a consensus document endorsed by the International Olympic Committee (6,37).
Incorporating ECG into a screening protocol improves efficacy in identifying conditions capable of causing SCD. It is the gold-standard investigation for detection of electrical abnormalities such as Wolff-Parkinson-White syndrome, and ion channelopathies, including LQTS and BrS. ECG is also effective in identifying cardiomyopathy, and its findings are abnormal in >90% of individuals with HCM and >75% of individuals with ARVC (18,66). The most compelling evidence in support of the Italian PPS model comes from a prospective study with 25 years of follow-up that compared the incidence of SCD in the pre-screening (1979 to 1982) and post-screening eras. This demonstrated a significant reduction in the incidence of SCD from 3.6/100,000 person-years to 0.4/100,000 person-years (p < 0.001), representing a 90% reduction in mortality (9). The predominant reason for this reduction was decreased SCD due to cardiomyopathy, in particular, ARVC, which was a relatively novel entity during the pre-screening era.
Despite such strong evidence for incorporating ECG in PPS, controversy remains, and the American Heart Association does not support routine use of ECG. The primary arguments against electrocardiographic screening include concerns regarding false-positive results, cost-effectiveness, and psychological implications for athletes and their families (5). Sudden death in the sports arena remains rare, and ECG cannot identify all conditions associated with SCD. Due to overlap between the physiological electrocardiographic changes seen in athlete's heart and similar changes seen in pathological states, it is important that evaluation is performed by highly trained cardiologists and sports physicians with expertise and experience in dealing with athletes and the complex phenotypic expressions of inherited cardiac diseases. The diversity in age, sex, ethnicity, and sport among the modern athletic population further complicates matters, particularly as the majority of published data originate from adult white males. Applying established guidelines for electrocardiographic abnormalities to athletes has been shown to generate false-positive rates between 4% and 7%, which has important implications for both the athlete and physician (67). However, the majority of SCD occurs in individuals without antecedent symptoms and with unremarkable cardiovascular examination, thus highlighting the advantages of evaluation incorporating ECG.
Studies comparing the cost-effectiveness of the U.S. and Italian screening models find in favor of the Italian protocol (68). However, the national screening program in Italy is already well organized and established. It may not be possible to reproduce these results in other countries with limited infrastructure to run a nationwide screening program and fewer trained medical personnel to implement it. An important question is where the financial burden of large-scale screening should fall; should it be paid for by sporting bodies and teams or regional and national governments? There are limited data evaluating the psychological implications of screening on athletes. A study of Norwegian soccer players showed the majority of individuals were satisfied having completed the screening and felt more confident. Less than 3% were distressed by it, and all the players involved would recommend screening to others (69). Data from other established screening programs, including prostate and breast cancer, demonstrate considerable anxiety generated by false-positive results until further investigations provide reassurance, highlighting the importance of prompt evaluation and referral to specialist centers (70). With athletes, there are significant social and financial losses to consider, as well as the long-term implications regarding family screening and insurance policies.
Automated external defibrillators
In the event of cardiac arrest in an athlete, survival is improved by prompt recognition, the presence of trained medical personnel to initiate cardiopulmonary resuscitation, and early access to an automated external defibrillator. Creation of emergency response plans at sports and athletic venues may improve the outcome of SCD events in athletes. A recent analysis of 1,710 U.S. high schools with on-site automated external defibrillators reported 36 cases of cardiac arrest. Of the 36 cases of cardiac arrest, a total of 23 individuals (64%) survived to hospital discharge, demonstrating that early defibrillation provides a survival benefit to young athletic victims of cardiac arrest (71).
Conclusions
SCD in the sports arena is rare but devastating. Preventive strategies, such as large-scale PPS of competitive athletes and increasing availability of automated external defibrillators are challenges requiring significant infrastructure and expertise but should be considered achievable aims rather than impossible goals. Victims of SCD are often entirely asymptomatic before their initial presentation and demonstrate only subtle abnormalities on investigation. It is therefore recommended that cardiac evaluation of an athlete is performed by trained cardiologists and sports physicians familiar with the conditions capable of causing SCD and the impact of demographic factors associated with the individual athlete.
Footnotes
Drs. Chandra and Papadakis are funded by research grants from the charitable organization Cardiac Risk in the Young (CRY), which supports pre-participation screening in young athletes. Prof. Sharma is consultant cardiologist to CRY and a CRY trustee. Ms. Bastiaenen has reported that she has no relationships relevant to the contents of this paper to disclose.
Abbreviations and Acronyms
ARVC: arrhythmogenic right ventricular cardiomyopathy
BrS: Brugada syndrome
CCAA: congenital coronary artery anomaly
ECG: electrocardiography
ER: early repolarization
ESC: European Society of Cardiology
HCM: hypertrophic cardiomyopathy
ICD: implantable cardioverter-defibrillator
LQTS: long QT syndrome
LV: left ventricular
LVH: left ventricular hypertrophy
LVWT: left ventricular wall thickness
MVP: mitral valve prolapse
PPS: pre-participation screening
RV: right ventricular
SCD: sudden cardiac death
VF: ventricular fibrillation
VT: ventricular tachycardia
Received May 28, 2012.
Revision received July 17, 2012.
Accepted August 13, 2012.
American College of Cardiology Foundation
References
- De Backer G., Ambrosioni E., Borch-Johnsen K., et al. (2003) European guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J 24:1601–1610.
- Corrado D., Basso C., Rizzoli G., Schiavon M., Thiene G. (2003) Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol 42:1959–1963.
- Sharma S. (2003) Athlete's heart—effect of age, sex, ethnicity and sporting discipline. Exp Physiol 88:665–669.
- Pelliccia A., Fagard R., Bjornstad H.H., et al. (2005) Recommendations for competitive sports participation in athletes with cardiovascular disease. Eur Heart J 26:1422–1445.
- Chandra N., Papadakis M., Sharma S. (2010) Preparticipation screening of young competitive athletes for cardiovascular disorders. Phys Sportsmed 38:54–63.
- Ljungqvist A., Jenoure P., Engebretsen L., et al. (2009) The International Olympic Committee (IOC) Consensus Statement on periodic health evaluation of elite athletes March 2009. Br J Sports Med 43:631–643.
- Corrado D., Pelliccia A., Bjornstad H.H., et al. (2005) Cardiovascular pre-participation screening of young competitive athletes for prevention of sudden death: proposal for a common European protocol. Eur Heart J 26:516–524.
- Maron B.J., Thompson P.D., Ackerman M.J., et al. (2007) Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update. Circulation 115:1643–1655.
- Corrado D., Basso C., Pavei A., Michieli P., Schiavon M., Thiene G. (2006) Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA 296:1593–1601.
- Drezner J.A., Rao A.L., Heistand J., Bloomingdale M.K., Harmon K.G. (2009) Effectiveness of emergency response planning for sudden cardiac arrest in United States high schools with automated external defibrillators. Circulation 120:518–525.
- Harmon K.G., Asif I.M., Klossner D., Drezner J.A. (2011) Incidence of sudden cardiac death in national collegiate athletic association athletes. Circulation 123:1594–1600.
- Van Camp S.P., Bloor C., Mueller F.O., Cantu R., Olson H. (1995) Nontraumatic sports death in high school and college athletes. Med Sci Sports Exerc 27:641–647.
- Pelliccia A., Maron B.J., Culasso F., Spataro A., Caselli G. (1996) Athlete's heart in women: Echocardiographic characterization of highly trained female athletes. JAMA 276:211–215.
- Chandra N., Papadakis M., Sharma S. (2012) Cardiac adaptation in athletes of black ethnicity: differentiating pathology from physiology. Heart 98:1194–1200.
- Maron B.J., Doerer J.J., Haas T.S., Tierney D.M., Mueller F.O. (2009) Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation 119:1085–1092.
- Maron B.J., Shirani J., Poliac L.C., Mathenge R., Roberts W.C., Mueller F.O. (1996) Sudden death in young competitive athletes; clinical, demographic and pathological profiles. JAMA 276:199–204.
- Maron B.J., Gardin J.M., Flack J.M., Gidding S.S., Kurosaki T.T., Bild D.E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 92:785–789.
- Maron B.J. (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320.
- Spirito P., Seidman C.E., McKenna W.J., Maron B.J. (1997) The management of hypertrophic cardiomyopathy. N Engl J Med 336:775–785.
- Garratt C.J., Elliott P., Behr E., et al. (2010) Heart Rhythm UK position statement on clinical indications for implantable cardioverter defibrillators in adult patients with familial sudden cardiac death syndromes. Europace 12:1156–1175.
- Basso C., Corrado D., Marcus F.I., Nava A., Thiene G. (2009) Arrhythmogenic right ventricular cardiomyopathy. Lancet 373:1289–1300.
- Corrado D., Basso C., Thiene G., et al. (1997) Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 30:1512–1520.
- Marcus F.I., McKenna W.J., Sherrill D., et al. (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur Heart J 31:806–814.
- Corrado D., Leoni L., Link M.S., et al. (2003) Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 108:3084–3091.
- Angelini P. (2007) Coronary artery anomalies: an entity in search of an identity. Circulation 115:1296–1305.
- Basso C., Maron B.J., Corrado D., Thiene G. (2000) Clinical profile of congenital coronary artery anomalies with origin from the wrong aortic sinus leading to sudden death in young competitive athletes. J Am Coll Cardiol 35:1493–1501.
- Prakken N., Cramer M., Olimulder M., Agostoni P., Mali W., Velthuis B. (2010) Screening for proximal coronary artery anomalies with 3-dimensional MR coronary angiography. Int J Cardiovasc Imaging 26:701–710.
- Yetman A.T., Bornemeier R.A., McCrindle B.W. (2003) Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol 41:329–332.
- Maron B.J., Zipes D.P. (2005) Eligibility recommendations for competitive athletes with cardiovascular abnormalities. J Am Coll Cardiol 45:1312–1375.
- Maron B.J., Ackerman M.J., Nishimura R.A., Pyeritz R.E., Towbin J.A., Udelson J.E. (2005) Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol 45:1340–1345.
- Kligfield P., Levy D., Devereux R.B., Savage D.D. (1987) Arrhythmias and sudden death in mitral valve prolapse. Am Heart J 113:1298–1307.
- Jeresaty R. (1986) Mitral valve prolapse: definition and implications in athletes. J Am Coll Cardiol 7:231–236.
- Heidbuchel H., Panhuyzen-Goedkoop N., Corrado D., et al., Study Group on Sports Cardiology of the European Association for the Cardiovascular Prevention Rehabilitation (2006) Recommendations for participation in leisure-time physical activity and competitive sports in patients with arrhythmias and potentially arrhythmogenic conditions: Part I: supraventricular arrhythmias and pacemakers. Eur J Cardiovasc Prev Rehab 13:475–484.
- Timmermans C., Smeets J.L.R.M., Rodriguez L.-M., Vrouchos G., van den Dool A., Wellens H.J.J. (1995) Aborted sudden death in the Wolff-Parkinson-White syndrome. Am J Cardiol 76:492–494.
- Cohen M.I., Triedman J.K., Cannon B.C., et al. (2012) PACES/HRS expert consensus statement on the management of the asymptomatic young patient with a Wolff-Parkinson-White (WPW, ventricular preexcitation) electrocardiographic pattern: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS): Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the American Academy of Pediatrics (AAP), and the Canadian Heart Rhythm Society (CHRS). Heart Rhythm 9:1006–1024.
- Schwartz P.J., Priori S.G., Spazzolini C., et al. (2001) Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103:89–95.
- Corrado D., Pelliccia A., Heidbuchel H., et al. (2010) Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J 31:243–259.
- Schwartz P., Moss A., Vincent G., Crampton R. (1993) Diagnostic criteria for the long QT syndrome: An update. Circulation 88:782–784.
- Schwartz P.J., Spazzolini C., Priori S.G., et al. (2010) Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Circulation 122:1272–1282.
- Heidbuchel H., Corrado D., Biffi A., et al., Study Group on Sports Cardiology of the European Association for the Cardiovascular Prevention Rehabilitation (2006) Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions: Part II: ventricular arrhythmias, channelopathies and implantable defibrillators. Eur J Cardiovasc Prev Rehab 13:676–686.
- Antzelevitch C., Brugada P., Borggrefe M., et al. (2005) Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111:659–670.
- Priori S.G., Napolitano C., Tiso N., et al. (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103:196–200.
- Maron B.J., Gohman T.E., Kyle S.B., Estes N.A.M., Link M.S. (2002) Clinical profile and spectrum of commotio cordis. JAMA 287:1142–1146.
- Link M.S., Wang P.J., Pandian N.G., et al. (1998) An experimental model of sudden death due to low-energy chest-wall impact (commotio cordis). N Engl J Med 338:1805–1811.
- Eckart R.E., Scoville S.L., Campbell C.L., et al. (2004) Sudden death in young adults: a 25-year review of autopsies in military recruits. Ann Intern Med 141:829–834.
- Basso C., Carturan E., Corrado D., Thiene G. (2007) Myocarditis and dilated cardiomyopathy in athletes: diagnosis, management, and recommendations for sport activity. Cardiol Clin 25:423–429.
- Dhar R., Stout C.W., Link M.S., Homoud M.K., Weinstock J., Estes N.A. III. (2005) Cardiovascular toxicities of performance-enhancing substances in sports. Mayo Clinic Proc 80:1307–1315.
- Maron B.J., Pelliccia A. (2006) The heart of trained athletes: cardiac remodelling and the risks of sports, including sudden death. Circulation 114:1633–1644.
- Pelliccia A., Maron B.J., Spataro A., Proschan M.A., Spirito P. (1991) The upper limit of physiologic cardiac hypertrophy in highly trained elite athletes. N Engl J Med 324:295–301.
- Crouse S.F., Meade T., Hansen B.E., Green J.S., Martin S.E. (2009) Electrocardiograms of collegiate football athletes. Clin Cardiol 32:37–42.
- Basavarajaiah S., Boraita A., Whyte G., et al. (2008) Ethnic differences in left ventricular remodeling in highly-trained athletes: relevance to differentiating physiologic left ventricular hypertrophy from hypertrophic cardiomyopathy. J Am Coll Cardiol 51:2256–2262.
- Papadakis M., Carre F., Kervio G., et al. (2011) The prevalence, distribution, and clinical outcomes of electrocardiographic repolarization patterns in male athletes of African/Afro-Caribbean origin. Eur Heart J 32:2304–2313.
- Popovic Z.B., Kwon D.H., Mishra M., et al. (2008) Association between regional ventricular function and myocardial fibrosis in hypertrophic cardiomyopathy assessed by speckle tracking echocardiography and delayed hyperenhancement magnetic resonance imaging. J Am Soc Echocardiogr 21:1299–1305.
- Van Driest S.L., Ommen S.R., Tajik A.J., Gersh B.J., Ackerman M.J. (2005) Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clinic Proc 80:463–469.
- Bauce B., Frigo G., Benini G., et al. (2010) Differences and similarities between arrhythmogenic right ventricular cardiomyopathy and athlete's heart adaptations. Br J Sports Med 44:148–154.
- Scharhag J., Thunenkotter T., Urhausen A., Schneider G., Kindermann W. (2010) Echocardiography of the right ventricle in athlete's heart and hearts of normal size compared to magnetic resonance imaging: which measurements should be applied in athletes? Int J Sports Med 31:58–64.
- Oxborough D., Sharma S., Shave R., et al. (2012) The right ventricle of the endurance athlete: the relationship between morphology and deformation. J Am Soc Echocardiogr 25:263–271
- Zaidi S., Sheikh N., Gati S., Ghani S., Howes R., Sharma S. (2011) High prevalence of modified task force criteria for arrhythmogenic right ventricular cardiomyopathy in healthy elite male athletes. Eur Heart J 32(Suppl):182–183.
- Basavarajaiah S., Wilson M., Whyte G., Shah A., Behr E., Sharma S. (2007) Prevalence and significance of an isolated long QT interval in elite athletes. Eur Heart J 28:2944–2949.
- de Noronha S.V., Sharma S., Papadakis M., Desai S., Whyte G., Sheppard M.N. (2009) Aetiology of sudden cardiac death in athletes in the United Kingdom: a pathological study. Heart 95:1409–1414.
- Rawlins J., Carre F., Kervio G., et al. (2010) Ethnic differences in physiological cardiac adaptation to intense physical exercise in highly trained female athletes. Circulation 121:1078–1085.
- Di Paolo F.M., Schmied C., Zerguini Y.A., et al. (2012) The athlete's heart in adolescent Africans: an electrocardiographic and echocardiographic study. J Am Coll Cardiol 59:1029–1036.
- Bastiaenen R., Behr E.R. (2012) Early repolarisation: controversies and clinical implications. Heart 98:841–847.
- Noseworthy P.A., Weiner R., Kim J., et al. (2011) Early repolarization pattern in competitive athletes: clinical correlates and the effects of exercise training. Circ Arrhythm Electrophysiol 4:432–440.
- Cappato R., Furlanello F., Giovinazzo V., et al. (2010) J wave, QRS slurring, and ST elevation in athletes with cardiac arrest in the absence of heart disease: clinical perspective. Circ Arrhythm Electrophysiol 3:305–311.
- Marcus F.I. (2000) Electrocardiographic features of inherited diseases that predispose to the development of cardiac arrhythmias, long QT syndrome, arrhythmogenic right ventricular cardiomyopathy/dysplasia and Brugada syndrome. J Electrocardiol 33(Suppl):1–10.
- Pelliccia A., Maron B.J. (1995) Preparticipation cardiovascular evaluation of the competitive athlete: perspectives from the 30-year Italian experience. Am J Cardiol 75:827–829.
- Wheeler M.T., Heidenreich P.A., Froelicher V.F., Hlatky M.A., Ashley E.A. (2010) Cost-effectiveness of preparticipation screening for prevention of sudden cardiac death in young athletes. Ann Intern Med 152:276–286.
- Solberg E.E., Bjornstad T., Anderson T., Ekeberg O. (2012) Cardiovascular pre-participation screening does not distress professional football players. Eur J Prev Cardiol 19:571–577.
- McNaughton-Collins M., Fowler F.J. Jr.., Caubet J.-F., et al. (2004) Psychological effects of a suspicious prostate cancer screening test followed by a benign biopsy result. Am J Med 117:719–725.
- Drezner J.A., Rogers K.J. (2006) Sudden cardiac arrest in intercollegiate athletes: detailed analysis and outcomes of resuscitation in nine cases. Heart Rhythm 3:755–759.
Story Credit: http://www.onlinejacc.org/content/61/10/1027

